MRCP revision battle 28.1: Haemochromatosis
MRCP revision battle 28.2: Syphilis
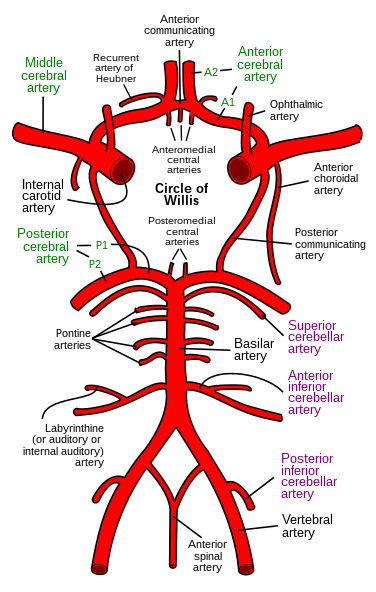
MRCP revision battle 28.3: Third nerve palsy
MRCP revision battle 28.4: G6PD deficiency
MRCP revision battle 28.5: Kaposi's sarcoma
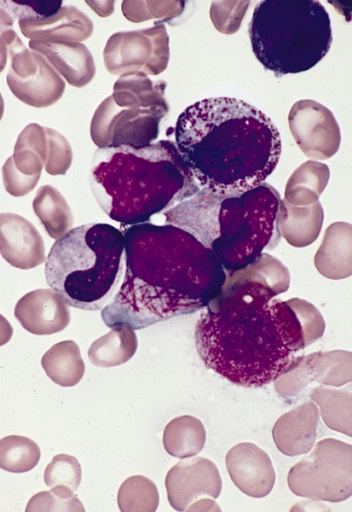
MRCP revision battle 28.6: Chronic Myeloid Leukaemia
MRCP revision battle 28.7: Gerstmann's Syndrome
MRCP revision battle 28.1: Haemochromatosis
Haemochromatosis is an excessive accumulation of iron.
There is increased intestinal absorption leading to deposition in joints, skin, the heart, liver, pancreas, adrenals and pituitary.
It is an autosomal recessive disorder, inherited on chromosome 6.
Presentation is initially with tiredness and arthralgia
Later problems include:
- diabetes mellitus (bronze diabetes)
- slate-grey skin
- liver disease/cirrhosis
- cardiac failure/cardiomegaly
- pseudogout
- hypopituitism
- >30% develop hepatocellular carcinoma
Males are affected earlier and more severely than females.
Diagnosis is by:
- transferrin saturation >50%
- ferritin >300-500micrograms/l (depending on guidelines)
- liver biopsy - Perls stain to show hepatic iron >180micromol/g
Treatment is regular venesection and ?chelation with desferrioxamine
Haemosiderosis is secondary haemochromatosis.
Causes of haemosiderosis include:
- beta thalassemia
- sideroblastic anaemia
- aplastic anaemia
- transfusions
- alcoholic cirrhosis
- chronic viral hepatitis
- porphyria cutanea tarda
Thats enough iron for one day... onwards to some syphilis...